
GxP Lifeline
Tips on How the TGA Regulates Drugs, Devices and Combination Products

Despite differences in application, development and regulatory pathways for medical firms with portfolios consisting of medicines, medical devices and biologicals/blood & tissue-based items (individually or in combination) all these products share common objectives in their use to test, investigate, diagnose, prevent, monitor, alleviate, compensate for, treat or cure a condition or disorder. Although most are easily identifiable as either a medicine or a medical device from its appearance, there are some that could be categorised either way.
The main difference is in how the product achieves its principal intended action/use. When the therapeutic effect is brought about by a pharmacological, chemical, immunological or metabolic means (i.e., via one or more ingredients with an active action), it is a medicine. If otherwise, e.g., mechanical, electrical or other action, the product is considered a medical device.
For example, saline (sodium chloride) solution may be considered a medicine or device depending on its intended use:
- For nasal irrigation (to clean and hydrate the nasal and sinus cavity) – medical device.
- For ophthalmic use (eye drops), nasal congestion/cough or as a diluent – medicine.
Other products may incorporate a device and medicine, with current regulatory pathways based on its primary function or use, for example, drug-coated coronary stents (device) and injectable medicinal solutions in syringes or auto-injectors (as a container). Immediate and future challenges for industry and regulators alike will be for medicine-device combinations when each has a synergistic contribution to the therapeutic benefit.
For now, separate regulatory guidelines and requirements are employed once a therapeutic product has been categorised as either a medicine or a device. However, these have common features despite differences in the details. In Australia, the following basic principles and concepts form the basis of Australia’s Therapeutic Goods Administration (TGA)’s oversight:
Risk Level The degree of documentation and data evaluated by the TGA depends on its benefit and safety risk:
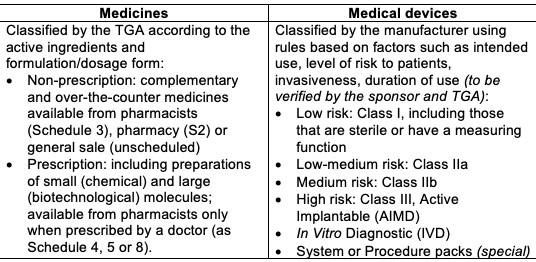
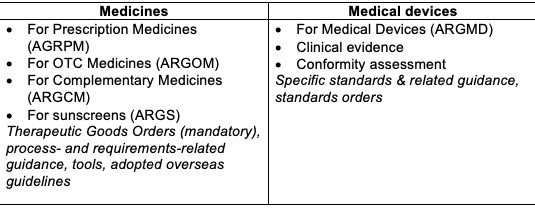
Australian regulatory guidelines include:
Development, Manufacturing and Quality Management
Both types of therapeutic goods are expected to be produced at a consistent and high standard of quality by manufacturers that employ a suitable quality management system (QMS) to oversee the product’s life cycle. Typical GxP standards adopted by the pharmaceutical industry include good manufacturing practice (GxP, which may vary slightly across different countries/jurisdictions), GCP (clinical), GLP (laboratory), GDP (documentation) while ISO 13485 and the ISO 9000 series are common QMS standards used by medical device manufacturers.
However, the level of QMS assessment and manufacturer authorization by the TGA differs between medical devices and medicines. For medical devices, a single legal manufacturer that is responsible for QMS oversight of its own manufacturing facilities and any subcontractors, design, development and compliance, is identified on the registration record. This manufacturer has an agreement with the local sponsor on responsibilities for post-marketing activities and regulatory maintenance.
In contrast, the manufacture of medicines involves several phases that may be performed by various manufacturers and facilities over an extended period of time. Since medicines principally rely on the activity and potency of one or more active ingredients, with other inactive or excipient ingredients combining to form the vehicle for drug delivery, the registration record identifies each manufacturing and control site responsible for:
- Starting materials for the drug substance, e.g., cell banks (for biotech medicines only).
- Drug substance (active) for chemical and biotech molecules (for prescription medicines only).
- Formulated product manufacture, packaging, testing and release.
Continuing authorization of the manufacturers by the TGA involves the periodic assessment of evidence for the device’s legal manufacturer and medicine’s various manufacturing sites as follows:
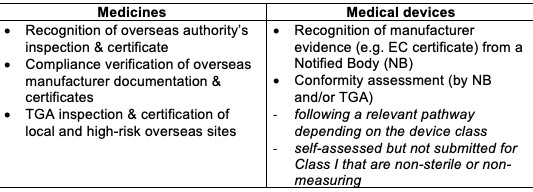
Keeping the manufacturer’s certification or clearance up-to-date is important to avoid delays in applications to vary registrations associated with the manufacturer or suspensions and cancellations of registration.
Regulatory Documentation/Dossier and TGA Evaluation
The level of evidence required for the evaluation of both product categories by the TGA is commensurate to the potential benefits and risks.
Information on the efficacy and safety of medicines are presented in a Common Technical Dossier (CTD) structure which consist of clinical, nonclinical (animal), quality, summaries and administrative information.
A technical file for medical devices is managed and maintained by the manufacturer to include information on design, development and manufacture, risk assessment, labelling/information, clinical evidence and post-marketing. Conformity with relevant Essential Principles is a key element and is often verified and certified by a Notified Body.
Registration Record (ARTG entry)A successful assessment results in entries in the Australian Register of Therapeutic Goods (ARTG):

Postmarket Responsibilities
After registration of both medicines and medical devices, the manufacturer’s and sponsor’s obligations include maintenance of the quality/design and labelling/user instructions information to ensure continuing effectivity and safety. Changes may be prompted by the manufacturer, from feedback by patients, health care professionals or users, or due to updates of standards and regulatory requirements. Some changes may be self-assessable, require notification to the TGA or must be approved prior to implementation depending on the nature of the change as described by the various regulatory guidelines.
After registration of both medicines and medical devices, the manufacturer’s and sponsor’s obligations include maintenance of the quality/design and labelling/user instructions information to ensure continuing effectivity and safety. Changes may be prompted by the manufacturer, from feedback by patients, health care professionals or users, or due to updates of standards and regulatory requirements. Some changes may be self-assessable, require notification to the TGA or must be approved prior to implementation depending on the nature of the change – as described by the various regulatory guidelines.
Other conditions of registration include pharmacovigilance, risk management, safety reports or alerts, management of complaints or failures, quality audits and traceability of the medicine batch or medical device unit. When necessary, both may be subject to recalls.

Vergel Manalo, team lead-medicines and senior consultant with Brandwood CKC, is qualified as a chemical engineer from St. Louis University in the Philippines, and recognized as a certified chemist (industrial) by the Royal Australian Chemical Institute (RACI) of Australia. He has more than 15 years’ experience in regulatory affairs involving registration and life cycle maintenance of prescription and non-prescription medicines, including new chemical and biological molecules, generics and biosimilars, over-the-counter and complementary products, and medicine-device combinations for the Australian and New Zealand markets. Manalo has extensive hands-on and management experience from over 20 years in the Australian and Asia-Pacific pharmaceutical industry in diverse roles, including manufacturing, quality, laboratory, validation, technical operations and product supply, and regulatory compliance.
FREE RESOURCE

Enjoying this blog? Learn More.
FDA and ISO Compliance for Medical Device Manufacturers
Download Now